# Bioinformatic Components
The 4 main sections of the pipeline are:
- Read alignment
- Somatic variant detection
- Germline variant detection
- Quality Ccontrol
Additionally, various QC metrics are generated. Below are described the separate modules tools used. The following diagram outlines the workflow:
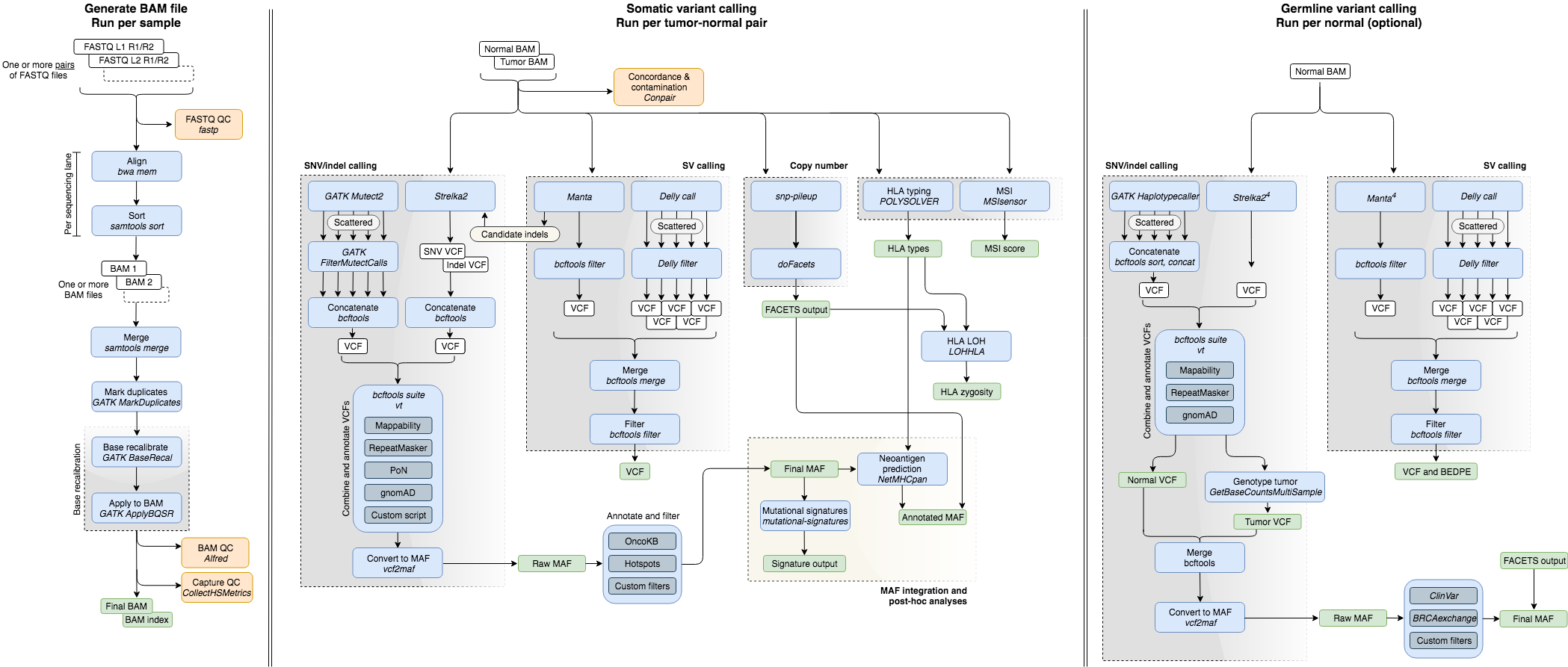
Note: The pipeline can be run with already-aligned BAM files as input, which avoids the first of these three modules.
# Read Alignment
Tempo accepts as input sequencing reads from one or multiple FASTQ file pairs (corresponding to separate sequencing lanes) per sample, as described. These are aligned against the human genome reference using common practices, which include:
- Alignment using BWA mem (opens new window), followed by conversion to BAM file format and sorting using samtools (opens new window).
- Merging of BAM files across sequencing lanes using samtools (opens new window).
- PCR-duplicate marking using GATK MarkDuplicates (opens new window).
- Base-quality score recalibration with GATK BaseRecalibrator and ApplyBQSR (opens new window).
# Somatic Analyses
- SNVs and indels are called using MuTect2 (opens new window) and Strelka2 (opens new window). Subsequently, they are combined, annotated and filtered as described in the section on variant annotation and filtering.
- Structural variants are detected by multiple callers and then merged, filtered and annotated as described in the section on variant annotation and filtering. Somatic and germline variants are generated with Delly (opens new window), Manta (opens new window) and SvABA (opens new window). Somatic whole genome variants are generated with the same callers, along with BRASS (opens new window).
- Copy-number analysis is performed with FACETS (opens new window) and processed using facets-suite (opens new window). Locus-specific copy-number, purity and ploidy estimates are integrated with the SNV/indel calls to perform clonality and zygosity analyses.
- Microsatellite instability is detected using MSIsensor (opens new window).
- HLA genotyping is performed with POLYSOLVER (opens new window).
- LOH at HLA loci is assessed with LOHHLA (opens new window).
- Mutational signatures are inferred with https://github.com/mskcc/tempoSig (opens new window).
- Neoantigen prediction using estimates of class I MHC binding affinity is performed with NetMHC 4.0 (opens new window) and integrated into the set of SNV/indel calls using https://github.com/taylor-lab/neoantigen-dev (opens new window) (Note: this repository is currently private).
- Clonality using Structural Variants is assessed using SVclone (opens new window). Joint calling is performed on both SNPs and structural variants, providing an alternative to the SNP clonality inference offered by Facets, while also assigning structural variants in the same clonal structure.
- Structural variant signatures are inferred with signature.tools.lib (opens new window)
- Homologous recombination deficiency is assessed using HRDetect from the signature.tools.lib package (opens new window)
# Germline Analyses
- SNVs and indels are called using HaplotypeCaller (opens new window) and Strelka2 (opens new window). Subsequently, they are combined, annotated and filtered as described in the section on variant annotation and filtering.
- Structural variants are detected by Delly (opens new window) and Manta (opens new window) then combined, filtered and annotated as described in the section on variant annotation and filtering.
# Quality Control
- FASTQ QC metrics are generated using fastp (opens new window).
- BAM file QC metrics are generated using Alfred (opens new window).
- Hybridisation-selection metrics are generated using CollectHsMetrics (opens new window). Only for exomes.
- Contamination and concordance metrics for tumor-normal pairs using Conpair (opens new window).
# MultiQC Report
A combined MultiQC report is produced at the BAM level, somatic level and cohort level of analysis, highlighting QC metrics and high-level summaries produced by Tempo. Documentation for the QC tool can be found here (opens new window).
The Tempo team has internally derived pass/warn/fail thresholds to present in the QC report. This should be used with discretion by the analyst, as a failure in the QC report may not be a true failure, and so on. By default the following metrics are assessed and a Status value is produced in the report:
- Tumor_Contamination : Estimates percentage of cross-individual contamination in the tumor sample. Cross-contamination may be higher if the sample comes from the recipient of allograft tissue.
- Normal_Contamination : Estimates percentage of cross-individual contamination in the normal sample. Cross-contamination may be higher if the sample comes from the recipient of allograft tissue.
- Concordance : measures likelihood of samples coming from the same individual. Low values may indicate sample swap or contamination.
- facets_qc and purity : Report by facets-preview (opens new window). A
facets_qcvalue ofFALSEindicates that the pair has failed Facets QC. - Fold Enrichment : The fold by which the baited region has been amplified above genomic background. A low metric could indicate inefficiency of the bait selection kit during sample preparation.
- __ Target Bases 50X__ : The fraction of all target bases achieving 50X or greater coverage. A low metric could indicate insufficient coverage.